

ABOUT PRIMARY HYPEROXALURIA TYPE 1 (PH1):
Il est normal d’avoir des questions
Peut-être vous sentez-vous submergé(e) d'avoir été diagnostiqué(e) ayant l'hyperoxalurie primitive de type 1 (HP1) et souhaitez en savoir plus sur cette maladie. Vous n’êtes pas seul(e). Voici les réponses à certaines questions courantes sur l’HP1:
Qu’est-ce que l’HP1 ?
L’hyperoxalurie primitive est une maladie héréditaire rare qui provoque une surproduction d’oxalate. Dans un foie sain, l’oxalate est présent uniquement en petites quantités. L’oxalate n’étant pas utilisé par l’organisme, il est éliminé par les reins.




Tour d’horizon sur l’HP1 : décrire l’HP1

L’HP1 entraîne une surproduction d’oxalate par le foie
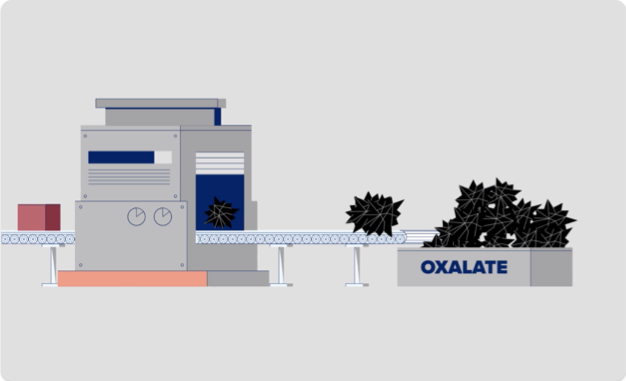
Dans l’HP1, l’oxalate est produit en excès en raison d’un processus défectueux qui implique des protéines (enzymes) hépatiques appelées glycolate oxydase (GO) et alanine-glyoxylate aminotransférase (AGT). Imaginez que votre foie est une usine dans laquelle ces enzymes travaillent comme des machines qui aideraient votre organisme à fabriquer ou à décomposer des substances. Voyez comment cela se produit ci-dessous:


Puisque l’HP1 est causée par une surproduction d’oxalate, votre médecin contrôlera les taux d’oxalate de votre organisme.





Que se passe-t-il en cas de surproduction d’oxalate?
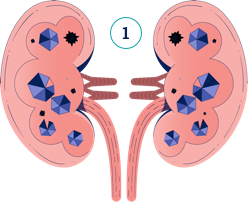
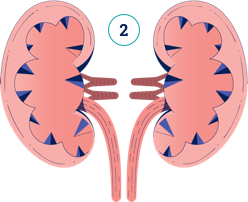
Dans l’HP1, l’oxalate est continuellement surproduit par le foie, et les reins ne parviennent pas à garder la cadence pour l’éliminer. Regardez la vidéo ci-dessous pour comprendre de quelle manière des dommages peuvent survenir.
Dans l’HP1, l’oxalate est continuellement surproduit et peut entrainer une aggravation de la maladie avec le temps. Plus le diagnostic est précoce plus il est facile de mettre en place les mesures nécessaires pour gérer la maladie.

Quels sont les signes à rechercher pour identifier l’HP1 ?
Bien que les calculs rénaux soient le signe le plus fréquent, et souvent le premier symptôme de l’HP1, tous les patients atteints d’HP1 n’en présentent pas nécessairement. Lorsque vous êtes atteint(e) d’HP1, vos reins risquent d’être endommagés même si vous ne développez pas de calculs rénaux.
L’HP1 peut se présenter de différentes manières :
Calculs rénaux,
même un seul, chez un enfant
Calculs rénaux à répétition
Présence de cristaux dans les tissus rénaux (néphrocalcinose), observée lors d’une exploration rénale
Insuffisance rénale terminale ou IRT
Retard de croissance (nourrisson)
Il y avait juste du sang dans ses urines.
Il ne
présentait aucun autre symptôme, mais l’on voyait qu’il souffrait. Je me souviens qu’il avait de nombreux calculs rénaux quand on l’a diagnostiqué.
Quels sont certains des symptômes que je pourrais rencontrer ?
Symptômes associés à la présence d’un calcul rénal (liste non exhaustive)
- Douleurs d'un côté du corps
- Douleurs lorsque vous urinez
- Sang dans les urines (hématurie)
- Infections des voies urinaires (IVU)
- Passage de calculs dans les urines
Si vous pensez présenter ces symptômes, songez à contacter votre médecin ou un urologue.
Symptômes associés à une insuffisance rénale terminale (IRT) (liste non exhaustive)
- Diminution ou arrêt de production de l’urine par les reins
- Sensation de malaise et de fatigue
- Perte d’appétit, nausées et vomissements
- Teint pâle
- Gonflement des mains et des pieds
- Dans l’HP1, une mauvaise fonction rénale peut occasionner la dispersion de l’oxalate dans l’organisme tout entier
Si vous pensez présenter ces symptômes, songez à contacter votre médecin ou un néphrologue. Grâce au suivi de votre fonction rénale, vous et votre médecin pouvez prendre des décisions plus éclairées quant à votre traitement.
Est-ce qu’un autre membre de ma famille peut également être atteint d’HP1 ?
L’HP1 est une maladie héréditaire, ce qui signifie qu’elle se transmet au sein des familles. Il est important que les membres de la famille d’une personne atteinte d’HP1, en particulier ses frères et sœurs, le cas échéant, envisagent d’effectuer un test génétique de dépistage de la maladie.


MON LIVRET HP1
Vous avez la possibilité de télécharger votre propre livret sur l'HP1, qui vous donnera un aperçu du suivi et de la prise en charge de l’HP1.

ENSUITE ?


Et ensuite ?
Vous et votre équipe médicale pouvez faire face à l’HP1 de différentes manières.
EN SAVOIR PLUS SUR LA PRISE EN CHARGEMilliner DS, et al. Primary Hyperoxaluria Type 1. 2002 Jun 19 [updated 2017 Nov 30].
Hoppe B, et al. Diagnostic and therapeutic approaches in patients with secondary hyperoxaluria. Front Biosci. 2003 Sep 1;8:e437-43.
Hoppe B, et al. The primary hyperoxalurias. Kidney Int. 2009 Jun;75(12):1264-1271.
Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012 Jun 12;8(8):467-75.
Cochat P, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012 May;27(5):1729-36.
Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013 Aug 15;369(7):649-58.
Ben-Shalom E, Frishberg Y. Primary hyperoxalurias: diagnosis and treatment. Pediatr Nephrol. 2015 Oct;30(10):1781-91.
Salido E, et al. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012 Sep;1822(9):1453-64.
Bhasin B, et al. Primary and secondary hyperoxaluria: Understanding the enigma. World J Nephrol. 2015 May 6;4(2):235-44.
Hopp K, et al. Rare Kidney Stone Consortium. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J Am Soc Nephrol. 2015 Oct;26(10):2559-70.
Edvardsson VO, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013 Oct;28(10):1923-42.
Falk N, et al. Primary hyperoxaluria type 1 with systemic calcium oxalate deposition: case report and literature review. Ann Clin Lab Sci. 2013 Summer;43(3):328-31.
Van Der Hoeven SM, et al. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant. 2012 Oct;27(10):3855-62.
Alelign T, Petros B. Kidney Stone Disease: An Update on Current Concepts. Adv Urol. 2018 Feb 4;2018:3068365.
Tang X, et al. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 2015 Mar;87(3):623-31.
OXL-FRA-00018 Avril 2021